Ketonuria ni uwepo wa mkojo wa miili ya ketone, ambayo ni acetone
Utaratibu wa ketonuria.
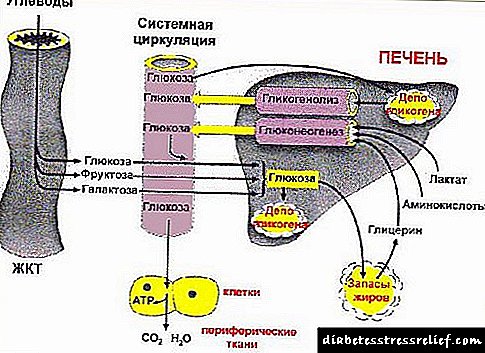
Katika mtu mwenye afya, wanga, lipids, na protini kadhaa hutiwa oksidi kaboni na maji. Katika hali zingine za patholojia, haswa na ugonjwa wa sukari, uzalishaji wa insulini hupungua. Katika ini, akiba ya glycogen hupunguzwa, tishu nyingi za mwili ziko katika hali ya njaa ya nishati. Chini ya hali hizi, michakato ya oxidation ya protini na lipids kwenye ini imeamilishwa, lakini ukosefu wa glycogen husababisha oxidation yao isiyo kamili na mkusanyiko katika damu ya bidhaa zilizo na athari ya metaboli ya lipid na proteni - miili ya ketone. Kwa sababu ya mkusanyiko wa miili ya ketone katika damu (ketonemia), pH ya damu huhamia upande wa asidi. Hali hii inaitwa acidosis. Mkojo wa mgonjwa kama huo una mmenyuko mkali wa asidi na harufu ya asetoni.
Kukabiliwa na njaa, matumizi ya vyakula vyenye protini na mafuta mengi, kutengwa kwa wanga kutoka kwa lishe husababisha kuongezeka kwa miili ya ketone na uchimbuaji wao katika mkojo.
Katika umri mdogo, ketonuria ni ya kawaida zaidi kuliko kwa watu wazima na haina umuhimu wa kliniki. Hali hii ni ya kupendeza kwa watoto na "kutapika kwa acetonemic" inayohusishwa na makosa katika lishe.
Ketonuria katika kipindi cha baada ya kazi ni kwa sababu ya kuvunjika kwa proteni kwa sababu ya kiwewe cha upasuaji.
Katika watu wazima, ketonuria hutokea katika aina kali za ugonjwa wa sukari na ni ya thamani kubwa ya utambuzi. Kwa watoto, inaweza kuwa na magonjwa anuwai, kwa sababu ya shida ya kimetaboliki ya wanga. Kwa hivyo, hata makosa madogo katika lishe, haswa mbele ya maambukizo ya papo hapo, msisimko wa neva, kazi ya kupita kiasi, n.k. inaweza kusababisha ketosis. Ketonuria katika utoto wa mapema inaweza kuzingatiwa na toxicosis, shida ya njia ya utumbo kwa muda mrefu, ugonjwa wa meno na magonjwa mengine. Katika watoto wachanga, kuongezeka kwa ketoni za mkojo karibu kila wakati husababishwa na utapiamlo. Ketonuria, inayozingatiwa katika magonjwa ya kuambukiza - homa nyekundu, homa, ugonjwa wa uti wa mgongo na ugonjwa wa ulevi, ni ishara ya sekondari, ni ya muda mfupi na haina thamani kubwa ya utambuzi.
Ketonuria ya ugonjwa wa sukari inakua kwa sababu ya kuongezeka kwa ketogene na ketolini iliyoharibika. Kuongezeka kwa ketogenesis husababisha kuongezeka kwa uhamishaji wa mafuta kutoka kwa tishu za adipose, kupungua kwa malezi ya oxalacetate katika mzunguko wa Krebs, na kupungua kwa biosynthesis ya asidi ya mafuta. Katika ugonjwa wa kisukari kali, unaongozana na uharibifu wa tishu za figo (mahali pa kuvunjika kwa ketoni), ukiukwaji wa ziada wa ketolysis hufanyika.
Kutokuwepo kwa glucosuria mbele ya ketonuria kuwatenga ugonjwa wa sukari.
Kawaida, miili ya ketone huundwa kwa kiasi kidogo kutoka kwa bidhaa ya mwisho ya kimetaboliki ya wanga na lipid - acetyl-CoA (C2-bodies) kupitia acetoacetyl-CoA na karibu kabisa haijachanganuliwa.
Katika ugonjwa wa kisukari mellitus, uhamasishaji wa lipids huongezeka na malezi ya idadi kubwa ya asidi ya acetyl (C2mwili). wakati huo huo, kwa sababu ya ukiukaji wa kimetaboliki ya wanga, kupungua kwa malezi ya oxalacetate, ambayo C2-kipimo ni pamoja na katika mzunguko wa Krebs na kuchoma hadi dioksidi kaboni na maji. Kwa kuongezea, kama matokeo ya kuongezeka kwa kuvunjika kwa lipid, biosynthesis ya nyuma ya C imefungwa.2mwili kuwa asidi ya mafuta. Kwa hivyo, idadi kubwa ya C kukusanya2-mtu kila mtu, ambayo husababisha uzalishaji wa idadi kubwa ya CoA, na kwa hivyo asetoni, asetacetiki na asidi ya beta-hydroxybutyric, ambayo hutiwa mkojo.
Miili ya Ketone
Hizi ni bidhaa za kuoka kati ambazo huundwa wakati wa kazi ya ini. Kawaida, kimetaboliki hufunua miili ya ketone kwa uharibifu zaidi.
Hifadhi za sukari mwilini hupatikana katika mafuta ya mwili, ndiyo sababu ketonuria ni ukosefu mkubwa wa wanga. Miili ya ketone ni mtoaji bora wa nishati. Katika hii, hata asidi ya mafuta iliyo nyuma yao. Kwa hivyo, wakati ubongo au moyo unakosa asidi ya fosforasi, mwili huzaa miili ya ketoni kwa kasi ya kasi.
Sababu ni zipi
Ketonuria ni yaliyomo ya mkojo wa miili ya ketone juu ya kawaida.
Katika mwili wa mwanadamu mwenye afya, kimetaboliki ina usawa. Ni nini kinachoweza kusababisha ketonuria?

- Umuhimu wa protini na mafuta katika lishe. Kwa sababu ya kupungua kwa wanga katika chakula, seli hukosa lishe. Kinyume na msingi huu, ketonuria inakua. Hii ndio majibu ya mwili kwa usawa katika lishe.
- Lishe na unyanyasaji wa njaa inaweza kusababisha kuonekana kwa miili ya ketoni kwenye mkojo. Makosa katika uteuzi wa lishe husababisha kupunguka kwa haraka kwa mafuta, wakati idadi ya Enzymes inaongezeka. Ketonuria pia ni muonekano wa asetoni katika damu. Kawaida, ikiwa mtu ana njaa kwa zaidi ya siku sita, yaliyomo kwenye miili ya ketone kwenye mwili wa binadamu huongezeka zaidi ya kawaida.
- Anesthesia ya upasuaji.
- Ugonjwa wa kisukari. Katika kesi hii, ketonuria inaonekana mara kwa mara. Miili ya Ketone pekee sio sababu ya ugonjwa. Katika ugonjwa wa kisukari mellitus, kozi ya ziada ya wanga na insulini imewekwa.
- Upungufu wa maji mwilini. Inatokea wakati mwili unapojaa au ni shida ya ugonjwa wa sukari.
- Ugonjwa wa ini au magonjwa hatari ya kuambukiza (k.v. Ugonjwa wa kuhara).
- Ukuaji wa tumors kwenye njia ya utumbo.
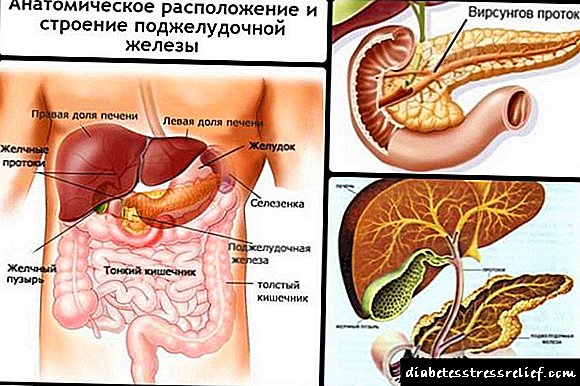
- Shida katika kongosho.
- Ku sumu kwa pombe au kwa misombo ya kemikali kama fosforasi, risasi.
- Uharibifu kwa mfumo mkuu wa neva, msisimko wa neva.
Kumbuka kuwa harufu ya asetoni wakati unapumua au mkojo ni ishara ya kumuona daktari. Pia ni sababu ya kubadilisha mtindo wa maisha na usawa.
Makini na watoto
Kliniki mara nyingi hushauriwa ikiwa mtoto ana kutapika, harufu ya acetone. Au harufu kama hiyo ilionekana kwenye mkojo. Na ingawa ni ishara ya ketonuria, sio lazima dalili ya ugonjwa mbaya.

Mara nyingi, kila kitu kinaelezewa na mfumo usio kamili wa metabolic. Sababu ya ketonuria kwa watoto ni hali ambayo nguvu kubwa hutolewa. Kawaida hutokea wakati:
- mzozo wa kihemko
- kuongezeka kwa nguvu ya mwili,
- lishe isiyo na usawa
- baridi
Ukweli ni kwamba mwili wa mtoto hauna maduka makubwa ya glycogen, kwa hivyo, kuvunjika kwa kazi kwa mafuta hufanyika na ishara za ketonuria huzingatiwa.
Mimba
Kwa miezi tisa ya ujauzito, mara nyingi wanawake wanapaswa kuchukua vipimo vya mkojo. Hii ni muhimu ili usikose kupotoka kidogo kutoka kwa kawaida katika mwili kwa kipindi chote cha ujauzito. Hakika, vyombo vyote vina mzigo mkubwa. Inamaanisha nini kwamba miili ya ketone hupatikana katika mkojo?
Kawaida yaliyomo kwao ni kawaida. Mchanganuo rahisi zaidi utakuwa kupita mtihani wa haraka. Unahitaji kupunguza kamba ya mtihani kwenye mkojo.
Mtihani hasi unachukuliwa kuwa wa kawaida wakati wa ujauzito au idadi ya ketoni ni ndogo. Ikiwa dhamana ya mtihani ni kutoka 15 hadi 160 mg / dl - hii ni sababu ya wasiwasi.
Wakati wa ujauzito, ketonuria ni ishara ya kutisha. Inatokea na toxicosis ya mapema. Kuumwa kwa mwili, na kwa hivyo kijusi na asetoni, huchanganya kozi ya ujauzito.

Ikiwa mwanamke mjamzito ana shida kubwa ya homoni, ana hatari.
Kuongezeka kwa kiwango cha ketoni kwenye mkojo wa mtoto mchanga ni kwa sababu ya ukosefu wa kutosha wa kulisha au makosa ya lishe.
Dalili za Ketonuria
Ikiwa kiwango cha ketoni mwilini kimeongezeka sana, basi ketonuria itajidhihirisha:
- uchovu mwingi
- harufu ya asetoni kutoka kinywani,
- harufu ya asetoni ya mkojo
- uchambuzi utaonyesha kiwango cha juu cha seli nyeupe za damu kwenye damu,
- mtihani wa damu ya biochemical utaonyesha kiwango cha chini cha sukari,
Ikiwa ketonuria ilisababisha kuruka katika damu ya asetoni, basi shida ya acetone inaweza kutokea.
Ongezeko kubwa la asidi katika seli zinaweza kuharibu vibaya viungo vya ndani. Katika kesi hii, mmenyuko wa kinga unazinduliwa - kutapika.
Dalili za ketonuria katika watoto:
- malalamiko ya maumivu ya tumbo
- malalamiko ya kichwa
- mtoto amechoka, amechoka,
- malalamiko ya kichefuchefu
- kutapika
- kuinua joto hadi 39 ° C,
- kukataa chakula
- harufu kama asetoni
- ini kubwa
Je! Ketonuria inaweza kugunduliwa?
Mchanganuo wa kemikali tu ndio unaoweza kuona kuongezeka kwa kiwango cha miili ya ketone mwilini. Maabara itaanzisha mara moja hali ya miili ya ketone iliyomo ndani yake.
Katika dawa ya kisasa, ketonuria hugunduliwa:
- Mtihani mkubwa,
- Mtihani wa kisheria
- Mfano Lestrade,
- mfano uliobadilishwa wa Rothera,
- vipimo vya haraka

Vipimo vya haraka, kwa kweli, ndio kawaida. Kitendo chao ni msingi wa mmenyuko wa kemikali, matokeo yake yanaonekana karibu mara moja. Mtu anapaswa kuweka tu kamba ya mtihani kwenye mkojo au aitupie kwenye kibao cha jaribio. Katika kesi ya mmenyuko mzuri, vipimo vinageuka zambarau. Mwangaza wa rangi hukuruhusu kuhukumu kwa kiwango maalum cha rangi juu ya jinsi kawaida ya miili ya ketone ilizidi.
Ainisho ya Kimataifa ya Magonjwa
Uainishaji wa Takwimu wa Kimataifa wa Magonjwa na Shida za kiafya zinazohusiana (ICD) ni mwongozo wa rejista ambayo vifaa vinaweza kulinganishwa kimataifa. Kwa msaada wake, njia anuwai za njia zimeunganishwa. Takwimu za ugonjwa zinakusanywa na kuainishwa. Kila miaka kumi, IBC inakaguliwa na tume ya Shirika la Afya Duniani. Ili kuainisha na kuchambua jumla ya matokeo yaliyokusanywa kutoka kwa mtazamo wa takwimu, hutafsiriwa kwa nambari za alphanumeric. Katika kazi kama hiyo, ICD iliyoendelea hutumiwa. Leo inalingana na ICD-10. Inayo madarasa 22 (sehemu).
Kulingana na saraka ya ICD, ketonuria ina nambari R82.4.
Kinga ya ketonuria na Lishe
Kwa kuzuia ketonuria, ni muhimu:
- kula sawa
- kuishi maisha ya afya:
- iwezekanavyo kuwa katika hewa safi,
- mazoezi ya wastani
- Usianze magonjwa sugu.

Katika ugonjwa sugu kama vile ugonjwa wa sukari, daktari anauliza kimfumo. Mara kwa mara ni muhimu kuchukua mtihani wa kuelezea.
Mwitikio mzuri unaonyesha ugonjwa wa mwili katika mwili. Zingatia ishara! Rufaa ya haraka kwa kliniki itarudisha mwili haraka kwa kawaida. Bila kuachana na lishe iliyowekwa na daktari, unaweza kuharakisha kupona.
Kanuni za chakula cha ketonuria:
- usila vyakula vyenye proteni na mafuta mengi,
- ulaji wa kawaida wa wanga
- kunywa maji zaidi na suluhisho la soda (na ugonjwa wa sukari - insulini).
Menyu inapaswa kujumuisha: nyama ya kuchemsha na sungura, supu za mboga anuwai, samaki wa chini-mafuta. Nafaka muhimu bila siagi, mboga na matunda. Inashauriwa kunywa juisi zaidi, vinywaji vya matunda, compotes.
Ondoa kutoka kwa menyu:
- nyama ya mafuta
- vitunguu saumu
- tamu
- matunda ya machungwa
- ndizi
- uyoga
- chakula cha haraka.
Zingatia matibabu
Ketonuria sio lazima kutibiwa kama ugonjwa tofauti. Yeye ni matokeo tu. Ni muhimu kuwatenga sababu zilizosababisha. Kwanza, uchunguzi kamili unahitajika. Usahihi wa utambuzi na uanzishwaji wa sababu ya ketonuria inahakikisha matibabu ya mafanikio.
Madaktari wanapeana vidokezo:
- Ikiwa wewe ni mzito, unapaswa kupanga siku za kufunga mara kwa mara.
- Ikiwa uchambuzi umebaini ongezeko la ketoni za mkojo, nunua vipimo ili utumie nyumbani.

- Mkojo unapaswa kukusanywa kwa uchambuzi si zaidi ya masaa manne kabla ya kujifungua.
- Mtoto anaweza kulewa na kinywaji cha alkali kila dakika 10-15 kwa sehemu ndogo. Mkaa ulioamilishwa, enterosgel itasaidia kusafisha njia ya utumbo.
- Kwa kutapika ujao, ni muhimu kuchukua kinywaji kibichi. Piga gari la wagonjwa.
Jinsi ya kutibu ketonuria, kuna vidokezo katika dawa za watu.
- Kwa kutapika mara kwa mara, kunywa maji ya madini, komputa wa matunda kavu, suluhisho la sukari. Kijiko moja katika dakika kumi.
- Weka enema nyumbani. Kwanza, maji kwenye joto la kawaida, joto baadaye, ambayo kijiko cha sukari huongezwa.
- Chukua kinywaji: kufuta 2 tbsp katika lita 1 ya maji. asali, mimina maji ya limao moja. Kunywa 1 tbsp. kila dakika 15.
- Kichocheo cha suluhisho la soda: kufuta kijiko 1 cha soda katika 250 ml ya maji. Kunywa 1 tsp. kila dakika 10.
- Chukua hatua za mimea ya kupendeza.
- Ili kuharakisha kuondoa kwa sumu kutoka kwa mwili, kula kidogo kidogo. Ni bora kula matapeli tu.
Matibabu inategemea tabia ya mtu binafsi ya mwili. Lazima iweze kudhibitiwa kabisa na daktari anayehudhuria.
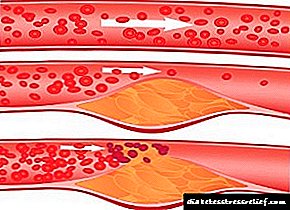
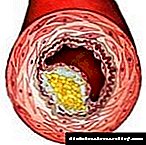
76. Cholesterol. Njia za kuingia, matumizi na excretion kutoka kwa mwili. Cholesterol ya Serum. Cholesterol biosynthesis, hatua zake. Udhibiti wa mchanganyiko.
Cholesterol ni steroid maalum kwa viumbe vya wanyama. Imeundwa ndani ya tishu nyingi za kibinadamu, lakini mahali pa msingi pa awali ni ini. Katika ini, zaidi ya 50% ya cholesterol imetengenezwa, ndani ya utumbo mdogo - 15-20%, cholesterol iliyobaki imechanganywa kwenye ngozi, kortini ya adrenal, na gonads. Karibu 1 g ya cholesterol imetengenezwa kwa siku katika mwili, 300-500 mg huingizwa na chakula (Kielelezo 8-65). Cholesterol hufanya kazi nyingi: ni sehemu ya membrane yote ya seli na inathiri mali zao, hutumikia kama sehemu ndogo ya awali katika muundo wa asidi ya bile na homoni za steroid. Watangulizi katika njia ya kimetaboliki ya awali ya cholesterol pia hubadilika kuwa ubiquinone, sehemu ya mnyororo wa kupumua na dolichol, ambayo inahusika katika awali ya glycoproteins. Kwa sababu ya kikundi chake cha hydroxyl, cholesterol inaweza kuunda ester na asidi ya mafuta. Cholesterol iliyo na mafuta huenea katika damu na huhifadhiwa kwa idadi ndogo katika aina fulani za seli ambazo hutumia kama sehemu ndogo ya muundo wa vitu vingine. Cholesterol na ekari zake ni molekuli za hydrophobic, kwa hivyo husafirishwa na damu tu kama sehemu ya aina tofauti za dawa. Kubadilishana kwa cholesterol ni ngumu sana - tu kwa muundo wake, karibu athari 100 mfululizo ni muhimu. Kwa jumla, karibu protini 300 tofauti zinahusika katika kimetaboliki ya cholesterol. Shida za kimetaboliki ya cholesterol husababisha moja ya magonjwa ya kawaida - atherosclerosis. Vifo kutoka kwa athari ya atherosclerosis (infarction ya myocardial, kiharusi) inaongoza katika muundo wa jumla wa vifo. Atherossteosis ni "ugonjwa wa polygenic", i.e. mambo mengi yanahusika katika maendeleo yake, ambayo muhimu zaidi ni ya urithi. Mkusanyiko wa cholesterol katika mwili husababisha ukuaji wa ugonjwa mwingine wa kawaida - ugonjwa wa gallstone.
A. Mchanganyiko wa cholesterol na kanuni zake
Athari za awali za cholesterol hufanyika katika cytosol ya seli. Hii ni moja ya njia ndefu zaidi ya kimetaboliki katika mwili wa binadamu.
Phenylketonuria

Phenylketonuria - ukiukwaji wa kurithi wa kimetaboliki ya amino asidi kutokana na ukosefu wa Enzymes ya ini inayohusika katika metaboli ya phenylalanine kwa tyrosine. Ishara za mwanzo za phenylketonuria ni kutapika, uchovu au shinikizo la damu, harufu ya ukungu kutoka kwa mkojo na ngozi, kucheleweshwa kwa maendeleo ya kisaikolojia, ishara za kawaida za marehemu ni pamoja na oligophrenia, kucheleweshwa kwa ukuaji wa mwili, kutetemeka, mabadiliko ya ngozi ya kizazi, nk Uchunguzi wa watoto wapya wa phenylketonuria unafanywa hata katika hospitali ya uzazi. Utambuzi uliofuata ni pamoja na uchunguzi wa maumbile ya maumbile, uamuzi wa mkusanyiko wa phenylalanine ya damu, uchambuzi wa biochemical, EEG, na MRI ya ubongo. Matibabu ya phenylketonuria ni kufuata lishe maalum.

Habari ya jumla
Phenylketonuria (Ugonjwa wa Felling, phenylpyruvic oligophrenia) ni ugonjwa wa kuzaliwa, na ugonjwa unaosababishwa na vinasaba unaodhihirishwa wa hydroxylation ya phenylalanine, mkusanyiko wa asidi ya amino na metabolites yake katika maji na tishu za kisaikolojia, ikifuatiwa na uharibifu mkubwa wa mfumo mkuu wa neva. Phenylketonuria ilielezewa kwa mara ya kwanza na A. Felling mnamo 1934; hutokea na mzunguko wa kesi 1 kwa watoto wapya 10,000. Katika kipindi cha neonatal, phenylketonuria haina dhihirisho la kliniki, hata hivyo, ulaji wa phenylalanine na chakula husababisha udhihirisho wa ugonjwa tayari katika nusu ya kwanza ya maisha, na baadaye husababisha shida kubwa ya maendeleo ya mtoto. Ndio sababu ugunduzi wa awali wa phenylketonuria katika watoto wachanga ni kazi muhimu zaidi ya neonatology, watoto na genetics.
Sababu za Phenylketonuria
Phenylketonuria ni shida ya urithi wa ugonjwa wa kurudia. Hii inamaanisha kuwa kwa ukuaji wa dalili za kliniki za phenylketonuria, mtoto lazima arithi nakala moja ya jeni kutoka kwa wazazi wote wawili, ambayo ni wabebaji wa heterozygous wa jenasi ya mutant.
Mara nyingi, maendeleo ya phenylketonuria husababishwa na mabadiliko katika gene iliyoingiza enzyme ya phenylalanine-4-hydroxylase iliyo kwenye mkono mrefu wa chromosome 12 (locus 12q22-q24.1). Hii ndio aina ya kinachojulikana ya classical I phenylketonuria, uhasibu kwa 98% ya kesi zote za ugonjwa. Hyperphenylalaninemia inaweza kufikia 30 mg% na ya juu. Ikiwa haijatibiwa, lahaja ya phenylketonuria hiyo inaambatana na kurudi nyuma kwa akili.
Mbali na fomu ya kitamaduni, phenylketonuria ya atypical inatofautishwa, ikiendelea na dalili sawa za kliniki, lakini haiwezi kurekebishwa na tiba ya lishe. Hii ni pamoja na aina ya phenylketonuria II (dehydroterterin reductase upungufu), aina ya phenylketonuria III (upungufu wa tetrahydrobiopterin) na zingine, tofauti adimu zaidi.
Uwezekano wa kuzaa mtoto na phenylketonuria huongezeka na ndoa za karibu.
Pathogenesis ya phenylketonuria
Njia ya classical ya phenylketonuria inatokana na ukosefu wa enzyme ya phenylalanine-4-hydroxylase inayohusika katika ubadilishaji wa phenylalanine kuwa tyrosine katika hepatocyte mitochondria. Kwa upande mwingine, tyramine inayotokana na tyramine ni nyenzo ya kuanzia ya awali ya katekisimu (adrenaline na norepinephrine), na diiodotyrosine kwa malezi ya thyroxine. Kwa kuongeza, malezi ya rangi ya melanin ni matokeo ya metaboli ya phenylalanine.
Upungufu wa kiini cha enzyme ya phenylalayin-4-hydroxylase katika phenylketonuria husababisha ukiukwaji wa oksidi ya phenylalanine kutoka kwa chakula, na kusababisha mkusanyiko wake katika damu (phenylalaninemia) na maji ya cerebrospinal huongezeka sana, na kiwango cha tyrosine hupungua ipasavyo. Phenylalanine ya ziada huondolewa na kuongezeka kwa mkojo wa mkojo wa metabolites yake - asidi ya phenylpyruvic, phenylmilactic na asidi ya phenylacetic.
Usumbufu wa kimetaboliki ya amino asidi unaambatana na ugonjwa wa kuvu wa nyuzi za ujasiri, kupungua kwa malezi ya neurotransmitters (dopamine, serotonin, nk), njia zinazosababisha pathogenetic ya kurudi nyuma kwa akili na shida ya akili inayoendelea.
Dalili za phenylketonuria
Watoto wachanga walio na phenylketonuria hawana ishara za kliniki za ugonjwa. Kawaida, udhihirisho wa phenylketonuria katika watoto hufanyika katika umri wa miezi 2-6. Kwa mwanzo wa kulisha, protini ya maziwa ya mama au viingilizo vyake huanza kuingia ndani ya mwili wa mtoto, ambayo husababisha maendeleo ya dalili za kwanza, zisizo maalum - uchovu, wakati mwingine wasiwasi na mshtuko wa hyper, regurgation, dystonia ya misuli, dalili ya kushtukiza. Moja ya ishara za mwanzo za pathognomonic ya phenylketonuria ni kutapika kwa kuendelea, ambayo mara nyingi hufikiriwa vibaya kama udhihirisho wa stenosis ya pyloric.
Kufikia nusu ya pili ya mwaka, mtoto aliyeko katika ukuaji wa kisaikolojia anaonekana. Mtoto huwa hafanyi kazi sana, hajali, huacha kugundua wapendwa, hajaribu kukaa chini na kusimama kwa miguu yake. Mchanganyiko usio wa kawaida wa mkojo na jasho husababisha harufu ya "panya" (harufu ya ukungu) inayojitokeza kutoka kwa mwili. Mara nyingi kuna ngozi ya ngozi, ugonjwa wa ngozi, eczema, scleroderma.
Katika watoto walio na phenylketonuria ambao hawapati matibabu, microcephaly, prognathia, baadaye (baada ya miaka 1.5) tething, hypoplasia ya enamel hugunduliwa. Ucheleweshaji wa maendeleo ya hotuba unajulikana, na kwa miaka 3-4 oligophrenia (idiocy) na kukosekana kabisa kwa hotuba hugunduliwa.
Watoto walio na phenylketonuria wana mwili wa dysplastiki, mara nyingi kasoro za moyo kuzaliwa, dysfunctions ya uhuru (jasho, acrocyanosis, hypotension ya zamani), na wanaugua kuvimbiwa. Tabia za phenotypic za watoto wanaougua phenylketonuria ni pamoja na ngozi nyepesi, macho na nywele. Mtoto aliye na phenylketonuria ni sifa ya tukio fulani la "Tailor" (miguu ya juu na chini ya miguu iliyoinama kwenye viungo), kutetemeka kwa mkono, mshtuko, nguvu ya nguvu, na hyperkinesis.
Dhihirisho la kliniki la aina II phenylketonuria linaonyeshwa kwa kiwango kikubwa cha kurudi kwa akili, kuongezeka kwa hasira, mshtuko, tetraparesis ya spastic, na hyperreflexia ya tendon. Kuendelea kwa ugonjwa kunaweza kusababisha kifo cha mtoto wa miaka 2 hadi 3.
Wakati aina ya phenylketonuri III inakua ishara ya ishara: microcephaly, oligophrenia, spetr tetraparesis.
Utambuzi wa phenylketonuria
Hivi sasa, utambuzi wa phenylketonuria (na galactosemia, hypothyroidism ya kuzaliwa, ugonjwa wa adrenogenital na cystic fibrosis) ni sehemu ya mpango wa uchunguzi wa neonatal kwa watoto wachanga wote.
Mtihani wa uchunguzi unafanywa kwa siku 3-5 ya maisha ya mtoto aliyemaliza kuzaa na siku 7 kwa kuchukua sampuli ya damu ya capillary kwenye fomu maalum ya karatasi. Ikiwa ugonjwa wa hyperphenylalanemia hugunduliwa, zaidi ya asilimia 2.2 ya mtoto hupelekwa kwa genetics ya watoto kwa uchunguzi upya.
Kuthibitisha utambuzi wa phenylketonuria, mkusanyiko wa phenylalanine na tyrosine katika damu hukaguliwa, shughuli ya enzymes ya hepatic (phenylalanine hydroxylase) imedhamiriwa, uchunguzi wa biochemical wa mkojo (uamuzi wa asidi ya ketoni), metabolites ya catecholamines kwenye mkojo, nk inafanywa na ugonjwa wa ubongo.
Kasoro ya maumbile katika phenylketonuria inaweza kugunduliwa hata wakati wa ujauzito wakati wa utambuzi wa ujauzito wa ujauzito wa fetusi (chorionbiopsy, amniocentesis, Cordocentesis).
Utambuzi tofauti wa phenylketonuria unafanywa na kiwewe cha ndani cha kuzaliwa kwa watoto wachanga, maambukizo ya intrauterine, na shida zingine za metabolic ya amino asidi.
Matibabu ya Phenylketonuria
Jambo la msingi katika matibabu ya phenylketonuria ni lishe ambayo inazuia ulaji wa protini mwilini. Matibabu inashauriwa kuanza katika mkusanyiko wa phenylalanine> 6 mg%. Mchanganyiko maalum umetengenezwa kwa watoto wachanga - Afenilak, Lofenilak, kwa watoto zaidi ya umri wa miaka 1 - Tetrafen, Phenyl-bure, zaidi ya umri wa miaka 8 - Maxamum-XP na wengine. Msingi wa lishe ni vyakula vyenye protini ya chini - matunda, mboga, juisi, hydrolysates za protini na mchanganyiko wa asidi ya amino. . Upanuzi wa lishe inawezekana baada ya miaka 18 kuhusiana na kuongezeka kwa uvumilivu kwa phenylalanine. Kwa mujibu wa sheria za Urusi, utoaji wa lishe ya matibabu kwa watu wanaougua phenylketonuria inapaswa kuwa bure.
Wagonjwa wameagizwa ulaji wa misombo ya madini, vitamini vya kikundi B, nk, kulingana na dalili - dawa za nootropic, anticonvulsants. Katika tiba tata ya phenylketonuria, massage ya jumla, tiba ya mazoezi, na acupuncture hutumiwa sana.
Watoto wanaosumbuliwa na phenylketonuria wako chini ya usimamizi wa daktari wa watoto na neuropsychiatrist, na mara nyingi wanahitaji msaada wa mtaalamu wa hotuba na daktari wa watoto. Uangalifu wa uangalifu wa hali ya neuropsychic ya watoto, ufuatiliaji wa kiwango cha phenylalanine katika damu na viashiria vya electroencephalogram ni muhimu.
Aina za kisayansi za phenylketonuria ambazo hazijaribika kwa matibabu ya lishe zinahitaji miadi ya hepatoprotectors, anticonvulsants, tiba mbadala na levodopa, 5-hydroxytryptophan.
Utabiri na kuzuia phenylketonuria
Kufanya uchunguzi wa wingi wa phenylketonuria katika kipindi cha neonatal hukuruhusu kupanga tiba ya mapema ya lishe na kuzuia uharibifu mkubwa wa ubongo, kuharibika kwa kazi ya ini. Kwa kuteuliwa mapema kwa lishe ya kuondoa kwa phenylketonuria ya classical, ugonjwa wa maendeleo ya watoto ni mzuri. Kwa matibabu ya marehemu, ugonjwa wa maendeleo ya akili ni duni.
Uzuiaji wa shida ya phenylketonuria ina uchunguzi wa watoto wachanga, kuagiza mapema na kufuata kwa muda mrefu na lishe ya lishe.
Ili kutathmini hatari ya kuzaa mtoto na phenylketonuria, ushauri wa maumbile ya maumbile unapaswa kutolewa kwa wenzi ambao tayari wana mtoto mgonjwa, wako kwenye uhusiano wa kuridhisha, na wana jamaa na ugonjwa huu. Wanawake walio na phenylketonuria ambao wanapanga ujauzito wanapaswa kufuata lishe kali kabla ya kuzaa na wakati wa ujauzito ili kuwatenga kuongezeka kwa kiwango cha phenylalanine na metabolites zake na ukuaji duni wa kijusi wenye afya ya kijenetiki. Hatari ya kupata mtoto na phenylketonuria katika wazazi wamebeba jeni lenye kasoro ni 1: 4.




-
Glycogen na kazi zake katika mwili wa binadamu
Glycogen kwenye mwili [hariri | kificho cha hariri] | hariri kanuni] Glycogen ni wanga ngumu ambayo ina safu ya masi ya sukari. ... -

-
-